Tu última visita fue el 05/05/2012 | 07:03:37
Búsqueda Simple
Búsqueda
Completa
Taxonómica
Anatomía

+ Teoría
Webs
Patología
Aleatorio Principio Activo/Comercial5 Fármacos o Drogas Más Visitadas
Desvenlafaxina

94054 visitas
Quetiapina

55797 visitas
Hidroxicina
55043 visitas
Clobenzorex
54249 visitas
Nicotina

43272 visitas
5 Últimos comentarios


Sigue los comentarios por

Total Fármacos Visitados
2.758.461
Desde Noviembre de 2008
Resolución fija 960px de ancho. Actualice navegador si todavía usa Internet Explorer 6
Nuestra WEB Amiga
CLASIFICACIÓN VARIOS 2


Nombre: Nalbufina
Comercial: Nubain, Bufilem, Fabitec
Foto: 
Fórmula: 
la nalbufina tiene una actividad mixta de agonista/antagonista de los receptores opioides. Estos incluyen los receptores µ (mu), k (kappa), y d (delta), que han sido recientemente reclasificados como OP1 (delta), OP2 (kappa), y OP3 (µ). La actividad de la nalbufina sobre los diferentes subtipos de receptores opioides se asemeja a la de la pentazocina, si bien la nalbufina es más potente con antagonista de los receptores OP3 y tiene menos efectos disfóricos que la pentazocina. Son los efectos agonistas de la nalbufina en los receptores k-1 y k-2 los que facilitan la analgesia. Se cree que los efectos sobre los receptores kappa producen alteraciones en la percepción del dolor así como en la respuesta emocional al mismo, posiblemente alterando la liberación de neurotransmisores desde los nervios aferentes sensibles a los estímulos dolorosos. Como adyuvante de la anestesia, se cree que la nalbufina protege el organismo frente a las agresiones ocasionadas por la cirugía. Paradójicamente, la nalbufina puede producir síndrome de abstinencia si se administra a pacientes opiáceo-dependientes. Este efecto es debido a un antagonismo sobre los receptores µ. Como la depresión respiratoria resulta de la estimulación de los receptores µ, la nalbufina produce menos depresión respiratoria que la morfina u otros agentes similares. Por el contrario, la nalbufina produce más efectos disfóricos que la morfina quizás debido a sus efectos estimulantes sobre los receptores sigma.
Fármaco consultado: 1919 veces
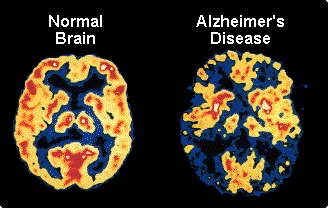
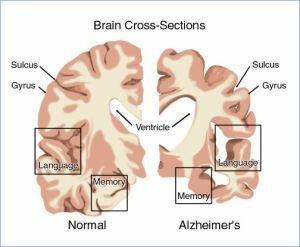
Enfermedad de Alzheimer - exploraciones PET muestran en los pacientes con Alzheimer leve y más en casos avanzados el metabolismo de glucosa se reduce tanto en los lóbulos temporal y parietal. Los pacientes con una mayor cantidad de disfunción del lenguaje, que problemas con la vista espacial muestran una reducción significativa en el metabolismo en el frontal izquierdo, lóbulos temporal y parietal. Los casos con mayor deterioro espaciales visuales tienen un metabolismo de la glucosa disminuida en el lóbulo parietal derecho.
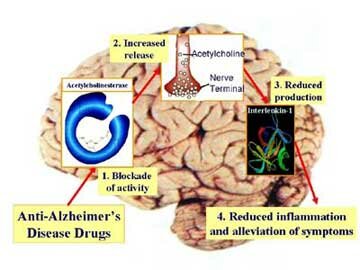
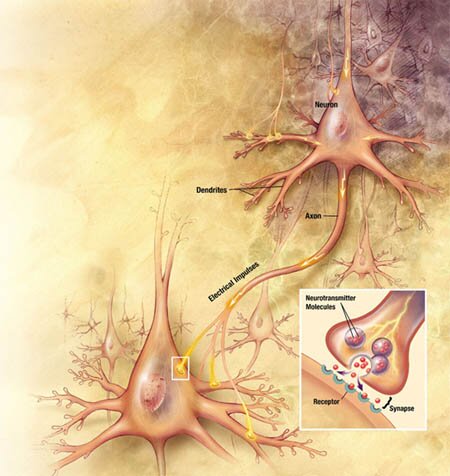
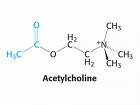
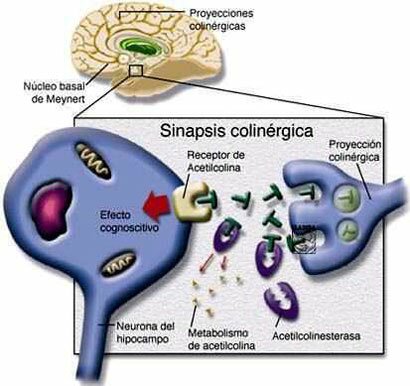
Inactivación de la Acetilcolina
El sistema de transporte vesicular de la Acetilcolina, responsable de la concentración de Acetilcolina en las vesículas sinápticas, ha sido caracterizado recientemente, a nivel molecular y funcional, e involucra un sistema torpedo electromotor especializado; la comparación del transporte de la Acetilcolina con los de las monoaminas demuestra la existencia de una nueva familia de genes; el mapeo de genes ha mostrado una única relación entre los genes para el transporte vesicular de la Acetilcolina y para la Colina-acetil-transferasa.
Una vez liberada a al hendidura sináptica, la acetilcolina se une durante un tiempo muy corto a sus receptores postsinápticos antes de ser degradada por la acetilcolinesterasa (AchE) que esta concentrada en la hendidura.
 La AchE es una glucoproteína globular que esta presente en los nervios, músculos y eritrocitos de los vertebrados y es especifica para la misma aceticolina. Esta enzima se sintetiza en el cuerpo celular y se distribuye a través de al neurona mediante flujo axoplasmico.
La AchE es una glucoproteína globular que esta presente en los nervios, músculos y eritrocitos de los vertebrados y es especifica para la misma aceticolina. Esta enzima se sintetiza en el cuerpo celular y se distribuye a través de al neurona mediante flujo axoplasmico.
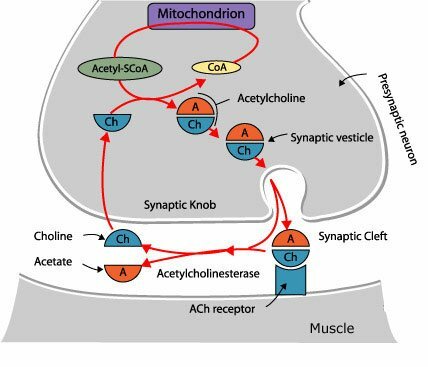
 La degradación hidrolítica de la Acetilcolina se lleva a cabo a nivel extracelular, en la proximidad de la terminación nerviosa, gracias a la acción de la Acetilcolinesterasa, que desdobla la Acetilcolina a sus componentes originales. Las aminas biogénicas en su mayoría son desactivadas por recaptación en las terminaciones nerviosas por las cuales fueron descargadas. Se ha demostrado la existencia de sistemas de captación altamente efectivos y específicos en todos los neurotransmisores del sistema nervioso central (Noradrenalina, Dopamina, Serotonina, GABA, Glutamato, Aspartato, Glicina), pero no para la Acetilcolina. Sin embargo, pese a que la Acetilcolina no tiene transportador, si existe un transportador de alta afinidad para su precursor, la Colina. Al parecer es probable que la desactivación por recaptura sea el mecanismo universal para la desactivación de las aminas y aminoácidos neurotransmisores; y que la degradación enzimática en el caso de la Acetilcolina es una excepción a la regla.
La degradación hidrolítica de la Acetilcolina se lleva a cabo a nivel extracelular, en la proximidad de la terminación nerviosa, gracias a la acción de la Acetilcolinesterasa, que desdobla la Acetilcolina a sus componentes originales. Las aminas biogénicas en su mayoría son desactivadas por recaptación en las terminaciones nerviosas por las cuales fueron descargadas. Se ha demostrado la existencia de sistemas de captación altamente efectivos y específicos en todos los neurotransmisores del sistema nervioso central (Noradrenalina, Dopamina, Serotonina, GABA, Glutamato, Aspartato, Glicina), pero no para la Acetilcolina. Sin embargo, pese a que la Acetilcolina no tiene transportador, si existe un transportador de alta afinidad para su precursor, la Colina. Al parecer es probable que la desactivación por recaptura sea el mecanismo universal para la desactivación de las aminas y aminoácidos neurotransmisores; y que la degradación enzimática en el caso de la Acetilcolina es una excepción a la regla.
La Acetilcolinesterasa se localiza primordialmente en neuronas colinérgicas (dendritas, pericariones y axones), en la proximidad de las sinapsis colinérgicas, y otros tejidos. De modo predominante, se localiza en las uniones neuromusculares, ganglios vegetativos, terminaciones nerviosas parasimpáticas y núcleo caudado. El plasma sanguíneo contiene un tipo inespecífico de la misma enzima conocido como Pseudocolinesterasa (Colinesterasa, Esterasa sérica o Butiril Colinesterasa)
Fuentes:
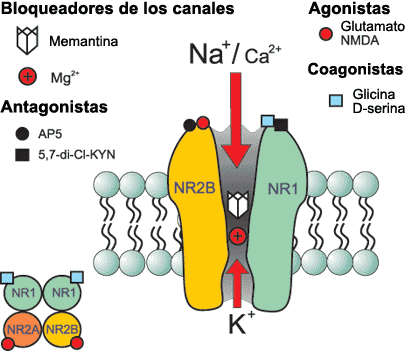
Hipótesis de trabajo en la búsqueda de nuevos fármacos
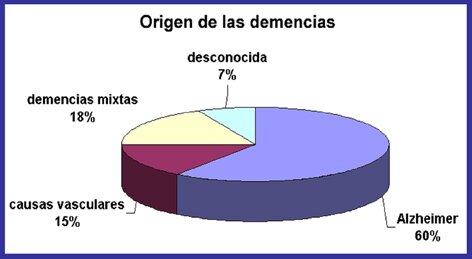
Las lesiones características de la enfermedad de Alzheimer se encuentran descritas desde principios del siglo XX y son las denominadas placas seniles, formadas por un péptido amiloide en un estado anómalo de agregación, y los ovillos neurofibrilares, que se localizan dentro de las neuronas formados por una proteina denominada tau en un estado aberrante de hiperfosforilación. Estas dos lesiones histopatológicas junto con la pérdida de las neuronas fundamentalmente colinérgicas, constituyen las bases moleculares de la enfermedad de Alzheimer. A partir de ellas, y con el fín de encontrar fármacos eficaces que acaben con esta enfermedad devastadora, se han originado tres hipótesis de trabajo:
ANTIDEMENCIALES:
HIPÓTESIS COLINÉRGICA:
En la que se intenta restaurar los niveles del neurotransmisor (acetilcolina) involucrado en los procesos de aprendizaje y memoria. Siguiendo estas ideas se inició una investigación farmacéutica intensa en la década de los 80 que concluyo con la aparición en el mercado de los actuales fármacos en uso como son tacrina, donepezilo, rivastigmina y galantamina. Son fármacos paliativos, pues mejoran la sintomatología del paciente sin llegar a interferir en el proceso neurodegenerativo.
En la enfermedad de Alzheimer, desde su inicio y a lo largo de la misma, falta en el cerebro una sustancia llamada acetilcolina, imprescindible para que el sistema de neuronas que la utiliza funcione normalmente trasmitiendo, a través de las sinapsis, los impulsos nerviosos por todo el circuito. Sin acetilcolina cerebral no hay memoria ni razonamiento. Por tanto, los enfermos van a mejorar si reciben agentes que les proporcionan esta sustancia. El donepezilo, la rivastigmina y la galantamina actúan en el cerebro mejorando la función de la acetilcolina. Todas las sustancias neurotrasmisoras que hay y se necesitan en el cerebro para un normal funcionamiento (como la dopamina, noradrenalina, serotonina, la susodicha acetilcolina, etc.) se han de sintetizar momento a momento, consumirse al realizar su misión de trasmitir impulsos acoplándose a las moléculas receptoras o receptores y, naturalmente, degradarse. Este un ciclo permanente y repetido diariamente. Esta degradación o metabolismo en el caso concreto de la acetilcolina la realiza una proteína o enzima llamada acetilcolinesterasa. La única manera que los científicos descubrieron hasta ahora de aumentar la acetilcolina en el cerebro de los enfermos con Alzheimer fue anular o inihibir la acción de este enzima. Por eso, los medicamentos que logran tal efecto se llaman anticolinesterásicos. Entre los tres medicamentos anticolinesterásicos disponibles hay diferencias en cuanto a su composición química, potencia, selectividad y modo de inhibir la acetilcolinesterasa, dosis, tiempo que permanecen en la sangre una vez absorbidos en el tubo digestivo, manera de viajar en la sangre para llegar al cerebro y mecanismo de eliminación del organismo. La rivastigmina tiene la peculiaridad de inhibir también otro enzima, llamado burtirilcolinesterasa, lo que puede conferirle un singular efecto clínico. Y la galantamina, además de inhibir, aunque sea débilmente la acetilcolinesterasa, destaca porque potencia la acción de los receptores neuronales a los que se acopla la acetilcolina, propiedad que la dota de un papel de protección neuronal.
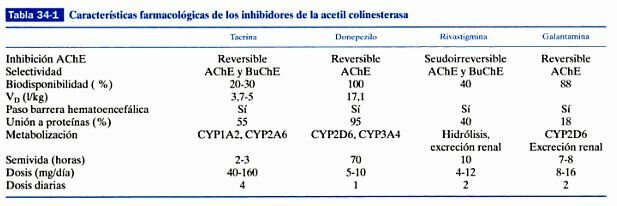
Fuente: Farmacología Humana Jesús Flórez.
HIPÓTESIS AMILOIDE:
Las investigaciones en este área se iniciaron una década mas tarde y actualmente hay diversos fármacos en diferentes estadios de desarrollo clínico. Se espera que a partir del 2011 se inicie la comercialización de alguno de ellos como fármacos modificadores del proceso neurodegenerativo. También, es importante señalar en este punto los diferentes estudios llevados a cabo con las técnicas de inmunización, activa y pasiva, o lo que es denominado como vacuna del Alzheimer. Aunque los primeros ensayos en pacientes tuvieron que ser suspendidos por la aparición de acontecimientos adversos graves, actualmente se han avanzado otras estrategias que los próximos años confirmaran o no las esperanzas depositadas en ellas.
HIPÓTESIS TAU:
Es la mas reciente de todas ellas, y sin embargo, al dia de hoy una de las mas prometedoras, pues se ha comprobado que el grado de demencia es directamente proporcional al número de ovillos neurofibrilares encontrados en los pacientes de Alzheimer y tambien de otras demencias denominadas tauopatias. Actualmente, un fármaco cuyo mecanismo molecular está basado en esta hipótesis se encuentra en desarrollo clínico con el fin de poder mostrar su eficacia en pacientes.
Dosis

Fuentes:
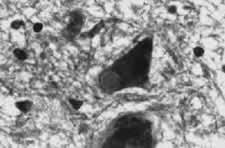
La demencia con cuerpos de Lewy (DCL) es una entidad clínico-patológica descrita recientemente. Sus principales características clínicas son el deterioro mental, parkinsonismo de intensidad variable, rasgos psicóticos como las alucinaciones visuales (AV), y fluctuaciones del estado cognitivo que afectan especialmente a la atención y concentración. Hasta la aparición en 1996 de los criterios diagnósticos del Taller Internacional del Consorcio para la Demencia con Cuerpos de Lewy, los casos de DCL solían ser erróneamente clasificados como enfermedad de Alzheimer (EA), enfermedad de Parkinson (EP) o la superposición de ambas. Aún hoy la DCL es infradiagnosticada clínicamente. Constituye más del 10% de todos los casos de demencia que llegan a la necropsia, pero los estudios de muestras clínicas informan que sólo alrededor de un 5% tienen el síndrome de DCL.
Inicialmente esta demencia suele ser diagnosticada como EA o DV pero, posteriormente y ante la presencia de síntomas parkinsonianos, el clínico la clasifica como DCL. En otros casos, los pacientes presentan inicialmente síntomas clásicos de la EP y más tarde desarrollan la demencia; solo una minoría de pacientes presenta inicialmente los síntomas de ambos trastornos.
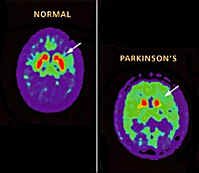
Enfermedad de Parkinson - Los pacientes tienen un mayor flujo al normal de sangre en los ganglios basales en el lado opuesto a las extremidades. L-dopa reduce el flujo de sangre a los ganglios de la base pero con el tiempo puede aumentar la tasa metabólica local. También hay una falta de comunicación entre el frontal y parietal y frontal con los lóbulos occipitales. Metabolismo de la glucosa también disminuyó en general en el cerebro sobre un 18% en comparación con una persona normal de la misma edad.
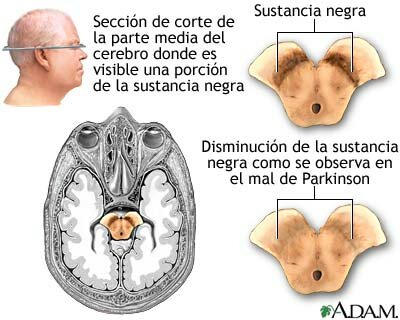
Es una enfermedad neurológica degenerativa que evoluciona a lo largo de los años y que suele aparecer en sujetos de edad avanzada. En la enfermedad de Parkinson la destrucción progresiva de una región específica del cerebro (la sustancia negra) desemboca en la aparición de síntomas cada vez más graves que pueden llegar a producir la muerte del paciente.
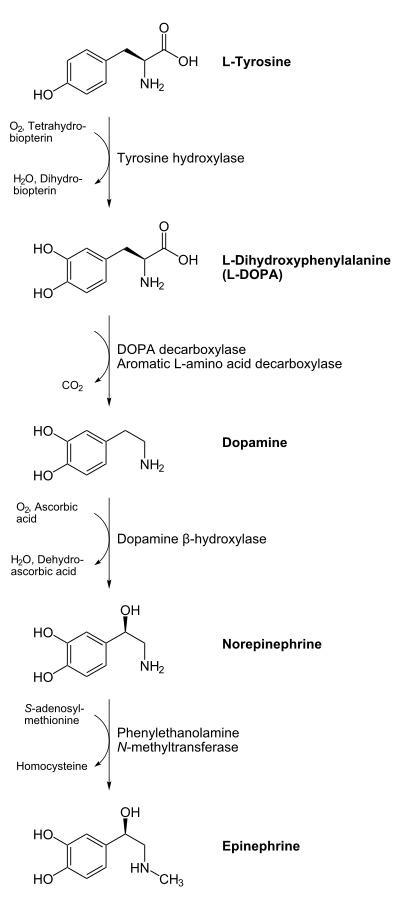
En el cerebro existe un grupo de células nerviosas encargadas de producir dopamina, un neurotransmisor esencial para el control de los movimientos y la transmisión del impulso nervioso. Estas neuronas se agrupan en una estructura denominada sustancia negra -por tener un color oscuro en los cadáveres-, que se sitúa en los ganglios basales.
Las neuronas de la sustancia negra de los sujetos con enfermedad de Parkinson mueren antes de tiempo sin ser sustituidas por otras nuevas. Cuando desaparece el 50 ó 60% de estas células de esta zona comienzan a hacerse evidentes los primeros síntomas: temblores, rigidez o dificultad para la marcha o el mantenimiento de la postura
LA ENFERMEDAD DE PARKINSON (EP) es un desorden neurodegenerativo de los ganglios basales (véase el capítulo II) que progresa hasta dejar al paciente incapacitado para efectuar movimientos voluntarios. Se observa aproximadamente en el 1% de la población mayor de 55 años, edad en la que la enfermedad es más frecuentemente diagnosticada. La EP se detecta primero como un ligero temblor en una o varias extremidades, rigidez muscular, anomalías posturales y lentitud de movimientos. Estos síntomas se pueden acompañar de pérdida del apetito y la subsecuente pérdida de peso, depresión, disminución de la capacidad intelectual y disfunciones autónomas (p. ejem., estreñimiento, seborrea, disfunción de la vejiga, hipotensión, sudoración excesiva, disfagia (dolor al deglutir), intolerancia al calor, alteraciones vasomotoras e impotencia sexual). La severidad de los síntomas puede aumentar en el transcurso de 10 a 15 años hasta reducir al paciente a un estado de completa inmovilidad y de demencia
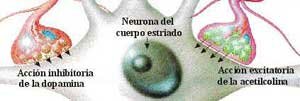
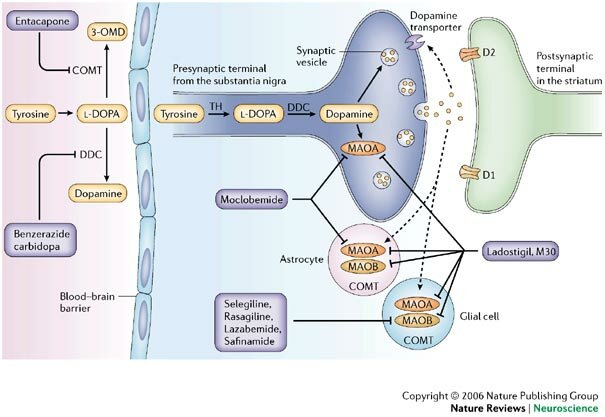
Durante aproximadamente 25 años el centro de la atención, en torno al tratamiento de la Enfermedad de Parkinson, estaba en el estriato (en realidad se refiere al neo estriato, es decir putamen y caudado pues es filogenéticamente más reciente que el paleo estriato o globus pallidus). En efecto se sabía que los neurolépticos bloqueaban los receptores dopaminérgicos post sinápticos a nivel del estriato causando un síndrome parkinsoniano que por esto no podía ser revertido administrando Levodopa. En el estriato había receptores D2 que inhibían la descarga post sináptica (al bloquear la enzima adenylciclasa) sobre los cuales se ejercía el efecto beneficioso de la Levodopa. Es decir la Levodopa es fundamentalmente un neurotransmisor depresor o bloqueador post sináptico. Además hay receptores D1 estriatales que, al contrario de los D2, aumentan la actividad post sináptica al incrementar la actividad de la enzima adenylciclasa y que contribuirían a la producción de las disquinesias. También sobre el estriato se efectuaban los transplantes de células productoras de dopamina obtenidas de la médula suprarenal, buscando ofrecer un mayor aporte de dopamina a los receptores post sinápticos.
Sin embargo en los últimos 7 a 10 años el centro de la atención está en el globus pallidus interno. Este, uno de los 2 núcleos del pallidus, al estar hiperactivo frena centros motores del tálamo y por consiguiente de la corteza cerebral premotora causando la sintomatología del Parkinson como temblor, hipoquinesia y aumento del tono muscular. Las conexiones entre estriato y globus pallidus interno son a través de una vía directa que inhibe este último núcleo teniendo así un efecto beneficioso en revertir la sintomatología Parkinsoniana. Esta vía cuyo neurotransmisor es GABA es estimulada por receptores D1 que son los que predominan. Hay otra vía estriato - pallidus interno que es estimuladora del globus pallidus interno y por lo tanto su hiperactividad es perjudicial. Esta vía es deprimida por los receptores D2. Esta vía es denominada indirecta pues del estriato pasa al núcleo pallidus externo donde hace sinapsis, posteriormente al núcleo subtalámico y de allí al núcleo pallidus interno. La dos primeras etapas tienen como neurotransmisores a GABA siendo inhibidores. La última del núcleo subtalámico a Pallidus interno es excitatoria siendo neurotransmisor el glutamato. El resultado final es que ejerce una acción activadora del núcleo pallidus interno. Por esto la Levodopa al actuar sobre receptores D1 y D2 ejerce una acción beneficiosa pues estimula la vía directa o inhibitoria y deprime la otra que es excitatoria (Gráfico 2).
Fuente:
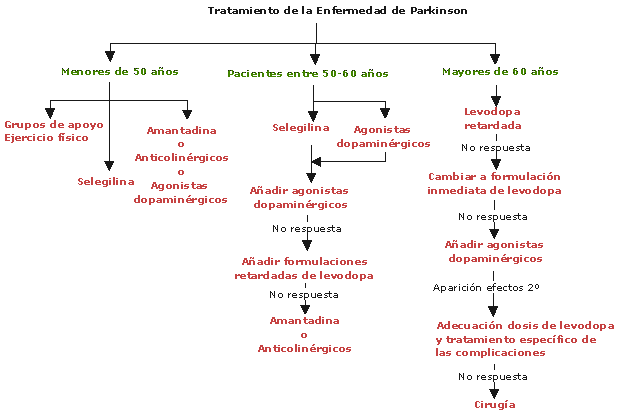
ANTIPARKINSONIANOS
Clasificación por Farmacodinamia:
AGONISTAS-DOPAMINA:
Favorecen la transmisión de dopamina estimulando los receptores dopaminérgicos. La mayoría de los enfermos necesitan asociar este tratamiento a la levodopa. En estudios de laboratorio se dice que podrían tener un efecto neuroprotector
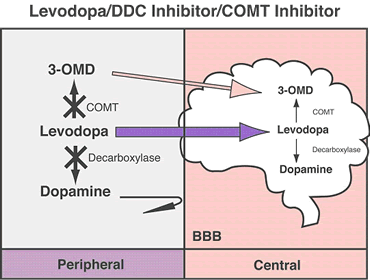
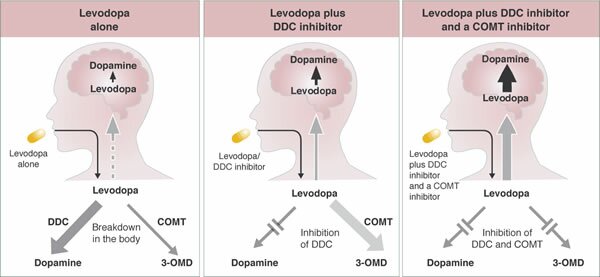
Inhibidor-DDC:
Los inhibidores DDC bloquean la enzima dopa-descarboxilasa periférica que causa que la levodopa descomponga la dopamina. Ellos se aseguran que más medicamento llegue a donde se necesita en el cerebro, lo cual también puede reducir algunos efectos secundarios potenciales, tales como nauseas y vómito. Estos efectos secundarios pueden ocurrir cuando la dopamina se encuentra en niveles altos en el torrente sanguíneo
Inhibidor-COMT:
La COMT (catecol-O-metiltransferasa) es una enzima que interviene en la metabolización de la levodopa antes de su entrada en el cerebro. Los inhibidores de esta enzima se utilizan para aumentar los niveles de levodopa que entran al cerebro y la disponibilidad de dopamina
ANTICOLINÉRGICOS:
Estructura, Aminas Terciarias. Restauran el equilibrio entre la dopamina y acetilcolina en los ganglios basales, bloqueando el exceso de acetilcolina, por la escasez de dopamina por utilización de neurolépticos o enfermedad de parkinson.

Fuente: Farmacología Humana Jesús Flórez.
Estudio comparativo de anticolinérgicos y placebo respecto al tratamiento de la acatisia
{kind=link}
Inhibidores de la MAO B(Selegilina): Ver Antidepresivos IMAOS B
Aumentan la disponibilidad de dopamina en el cerebro ya que reducen su degradación. Puede aumentar el tiempo 'on' y disminuir el tiempo 'off' en pacientes fluctuantes y ayuda a reducir las dosis totales de levodopa o el número de tomas.
Fuentes:
ANTICOREICOS
Utilizados para discinesias tardías, corea de huntington, balismo
Inhibidores del transportador vesicular VAMT:
Depleción de las monoaminas en el citoplasma por inhibición del transportador vesicular que lleva las monoaminas hasta las terminales presinápticas,
Fuentes:
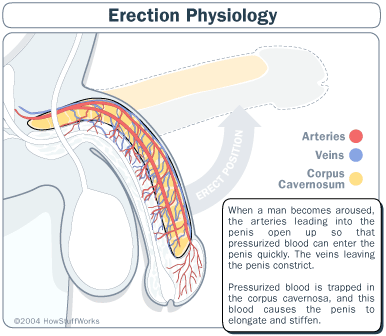
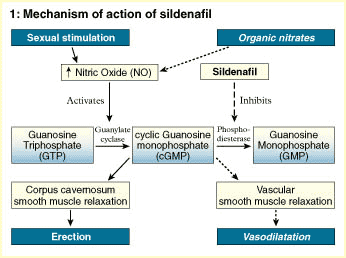
ANTI DISFUNCIÓN ERÉCTIL:
El citrato de sildenafil es una droga que suministrada por vía oral posee un efecto de inhibición marcada de la fosfodiesterasa específica tipo 5 (PDE5) que es una enzima predominante en el cuerpo cavernoso de los humanos. Consecuentemente tiene el potencial de ser efectivo en el tratamiento de la disfunción eréctil ya que Sildenafil (Viagra) aumenta el efecto de sustancias vasodilatadoras en los cuerpos cavernosos del pene. Una enzima específica es la responsable de la degradación de ese vasodilatador. Por ello un inhibidor selectivo de esa enzima facilita la relajación producida por el óxido nítrico en el músculo liso de los cuerpos cavernosos y aumenta la respuesta erectiva.
Otro mecanismo de amplificación es el implicado en el fenómeno de erección del pene, los científicos determinaron que en este caso el ligando es el NO (oxido nítrico), que es el principal neurotransmisor en el pene . En este caso, a diferencia de los descriptos, el receptor es el hierro del grupo hemo de la enzima guanilato-ciclasa y el segundo mensajero es el GMPc (Guanosin-Monofosfato-ciclico) en vez del AMPc. Una fosfodiesterasa (la isoenzima tipo V es la predominante), esta implicada en la inactivación del GMPc y por ende en la finalización de su efecto. La inhibición de esta fosfodiesterasa es la base del efecto del fármaco.
Fuente:
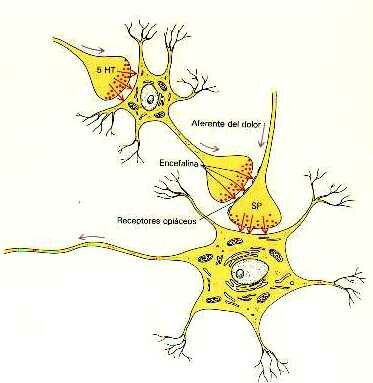
La existencia de estos receptores específicos de opiáceos en el sistema nervioso quiere decir que el organismo debe tener sus propios opiáceos. Efectivamente así es. Estos opiáceos naturales se denominan encefalinas o endorfinas. En esta figura vemos como la una neurona libera al final de su axón encefalina. Esta substancia se une o acopla a los receptores de opiáceos del axón o fibra nerviosa que conduce el dolor para bloquear la transmisión de las sensaciones dolorosas.
Fuente:
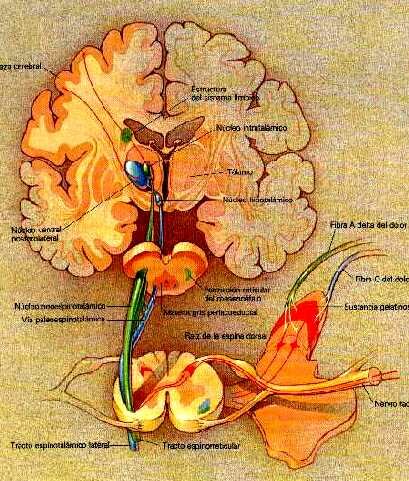
Las substancias derivadas del opio(morfina,heroina) ejercen su acción tanto de alivio del dolor como la euforia que producen al unirse o acoplarse a unos receptores específicos de opiáceos que se hallan ampliamente distribuidos por el cerebro tal como muestra esta figura.El sistema límbico, que como hemos reiteradamente indicado regula y controla nuestras emociones, es rico en estos receptores de opiáceos.

OPIÁCEOS: Nalbufina
Definición y descripción de las propiedades de algunos opiáceos y opioides
El presente resumen, extraído del libro de Seidenberg & Honegger (2000), pretende ser un texto orientativo para comprender mejor los programas de mantenimiento con opiáceos y opioides.
Los opiáceos son conocidos desde hace mucho tiempo como sustancias naturales, que se encuentran en el zumo de las semillas de la adormidera o papaver somniferum. El zumo seco y fermentado se denomina opio y contiene una mezcla de alcaloides opiáceos. En 1806, el químico alemán Fiedrich Serturner consiguió aislar el principal elemento del opio en su forma pura y que llamó morfina. Tras mínimas alteraciones químicas se pudieron obtener opiáceos semi-sintéticos. Desde hace 50 años, es posible obtener substancias completamente sintéticas, casi sin relación química con la morfina, pero con el mismo efecto.
A menudo se utiliza el término opiáceo en vez de opioide. No obstante el término opiáceo se refiere al origen de la sustancia con respecto al opio, es decir, son sustancias que se extraen de la cápsula de la planta del opio. Por extensión, se denominan también así los productos químicos derivados de la morfina. El término opioide se utiliza para designar aquellas sustancias endógenas o exógenas que tiene un efecto análogo al de la morfina y poseen actividad intrínseca. No todos los opioides son opiáceos, ni todos los opiáceos son opioides
Hughes describió en 1975 los péptidos del cuerpo humano que tenían efectos similares a la morfina. Los opioides actúan como éstos péptidos endógenos, denominados también endorfinas. Los péptidos opioides endógenos y las moléculas opioides, ya sean como medicina o como droga, reaccionan en la misma posición receptora específica en la superficie de las células nerviosas y de las células del músculo liso del intestino.
Los ligandos de receptores opioides son sustancias que se unen específicamente a los receptores opioides. Una vez unidos al receptor desarrollan un efecto que se denomina actividad intrínseca, y por esta razón son sustancias denominadas agonistas. Los ligandos de receptores opioides que se unen al receptor, pero no desarrollan efecto alguno, carecen de actividad intrínseca y se denominan antagonistas opioides.
| Opiáceos | Tipo | Características | Actividad Intrínseca | |
| Morfina |
§ Analgésico § Duración del efecto: 4-5 horas § Vida media: 3 horas |
Agonista | ||
| Codeína |
§ Suprime la tos § Duración del efecto: 4-6 horas § Vida media: 3-4 horas |
Agonista | ||
| Tebaína | § Sustancia a partir de la cual se sintetizan la naloxona, naltrexona, y buprenorfina | Agonista parcial | ||
| Papaverina | § Espasmolítico | Antagonista | ||
| Noscapina |
§ Suprime la tos sin adicción potencial
|
Antagonista | ||
| Opioides | Semi-sintéticos |
Heroína, (3, 6 diacetylmorphina, diamorfina DAM) |
§ Analgésico obtenido de la morfina § Duración del efecto: 4-5 horas § Vida media: media hora |
Agonista |
| Buprenorfina |
§ Analgésico § Inhibidor de la abstinencia § Duración del efecto: 6-8 horas § Vida media: 5 horas |
Agonista parcial | ||
| Sintéticos | Metadona |
§ Analgésico § Inhibidor de la abstinencia § Duración del efecto: 8-48 horas § Vida media: 15-22 horas |
Agonista parcial | |
Los opioides naturales y sintéticos, así como los péptidos opioides endógenos, se unen específicamente y con gran afinidad a los receptores opioides, lo que quiere decir que estas sustancias se acoplan perfectamente con los receptores opioides. Los receptores opioides se localizan frecuentemente en la porción final del axón presináptico de la célula nerviosa y modulan la liberación de los neurotransmisores al inhibir la entrada en funcionamiento del potencial de acción, con lo que disminuye la cantidad de sustancia transmisora liberada. El efecto de este receptor opioide es muy marcado en las células nerviosas que transmiten el dolor, donde la liberación de las sustancia transmisora del dolor o sustancia P se inhibe, lo que explica el efecto analgésico sobre los transmisores receptores opioides. Los diferentes opioides se unen con más o menos fuerza a los diferentes tipos de receptores de opioides: mu (m), delta (d) y kappa (k). Los opioides preferidos por los adictos y con un mayor efecto analgésico son los actúan particularmente en los receptoresm
Los opiodes exógenos presentan el llamado dualismo farmacológico. Este fenómeno consiste en que dos fármacos opioides actuando sobre receptores distintos (mu, delta, kappa) ejercen el mismo efecto farmacológico, por ejemplo, analgesia. Pero estos mismos fármacos pueden actuar como agonista en un receptor y agonista parcial o antagonista sobre el otro, siendo el resultado de su interacción distinto, como se muestra en la siguiente tabla: .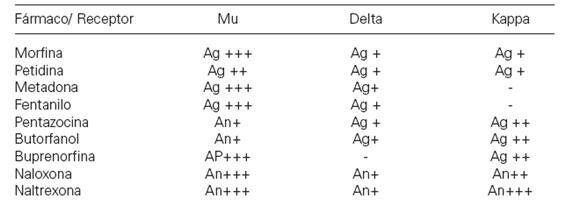
La actividad intrínseca relativa (AIR) se define como la capacidad de los ligandos para producir un efecto determinado en un receptor. La morfina y la metadona tienen la misma especificidad por el receptor opioide y como agonistas puros tienen el valor máximo de AIR. En principio producen el mismo efecto. Las diferencias subjetivas en lo que se refiere a su eficacia, se explican a través del comportamiento farmacocinético que es distinto para cada sustancia, en la capacidad de atravesar la barrera hemotoencefálica, en la distribución por los diferentes compartimentos (sangre, tejido cerebral, órganos internos) y finalmente en el metabolismo y la excreción.
Cuando hablamos de farmacocinética, nos referimos al transcurso de la concentración, es decir, variaciones de las concentraciones plasmáticas en función del tiempo, en el lugar del efecto. El curso temporal de la concentración depende de la absorción de la sustancia, la distribución en el organismo, el metabolismo y la eliminación. La absorción y la distribución determinan la presencia del fármaco (invasión o flooding) en el lugar del efecto. El abandono del fármaco del lugar de acción (evasión) está prácticamente determinado por el metabolismo y la eliminación.
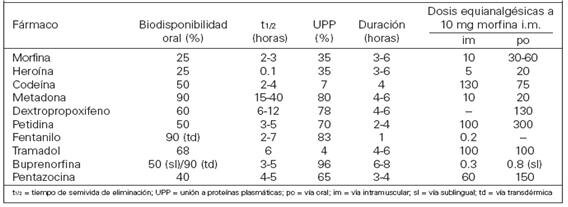
Por vía oral, la mayoría presenta una baja biodisponibilidad (<50%) debido a metabolismo de primer paso hepático. Tras su absorción, se distribuyen rápidamente en el organismo, variando su volumen de distribución entre 1.5 y 4.7 L/kg.
El mecanismo principal de inactivación es el metabolismo hepático que, suele consistir en una oxidación microsomal y la conjugación con ácido glucurónido. La desmetilación mediante el sistema encimático del citocromo P450 2D6, el citocromo (CYP2D6) es relevante en el metabolismo de la codeína, el tramadol y el dextrometorfano. La heroína se desacetilación por esterasa plasmática y la carboxilesterasa hepática en dos pasos, dando en el primero el metabolito 6-Monoacetilmorfina (6-MAM) y en el segundo la molécula de la morfina, parece ser que los efectos de la heroína de deben a la 6-MAM y la heroína que resultan de su metabolismo.Fuentes:
RECEPTORES Y OPIOIDES ENDOGENOS Y EXÓGENOS
INTRODUCCION :Hasta ayer se llamaba OPIOIDES a los agonistas endógenos de los receptores opiáceos y OPIACEOS a los agonistas exógenos.
Hoy se los denomina opioides endógenos y opioides exógenos respectivamente.
RECEPTORES- HISTORIA
Existen una serie de evidencias que avalan el concepto de Receptor: (ver diapo 1)1: La morfina es un alcaloide proveniente de una planta el papaver somniferum
2: Existe una similitud de fórmula química entre los distintos opioides como por ej. existen - amplias superficies lipofílicas en ángulo recto una respecto a la otra
un OH que se une por su H.
un N + que forma un puente iónico.
3. : Existe estereoisomería: el isómero levógiro es el farmacológicamente activo
mientras que el isómero dextrógiro no es activo salvo el dextrometorfan que es
antitusígeno y hoy se sabe que es un antagonista no competitivo del receptor NMDA.
4: Existe un antagonista para los opiodes que es la NALOXONA que revierte rápidamente la acción farmacología de los opioides
En 1967 Martín y colab. postularon la existencia de más de un tipo de receptor para los opioides y drogas afines, por las complejas interacciones de drogas del tipo de la Morfina, los antagonistas y lo agonistas - antagonistas.
En 1973 Snyder y Pert, Simon y Edelman, y Terenius, describieron separadamente sitios de unión estereoespecíficos saturables para los opioides y drogas afines.
En 1976 Martin y colab. descubrieron muchas categorías de receptores y los clasifica .
DESARROLLO DEL TEMA
CLASIFICACIÓN DE LOS RECEPTORES
(Ver diapo 2)Existen tres tipos de receptores: Mu, delta y kapa.
Hoy se sabe que hay 3 categorías de receptores opioides:
Mu1 y Mu2
K1a, 1b,2ª,2b
Delta 1 y delta 2
Epsilon, para el sistema inmunitario
. El receptor epsilon no se considera receptor opioide sino que está involucrado al sistema inmunitario.El receptor mu y el delta poseen un peso molecular entre 150 .000 y 750-875.000.
El receptor kappa, posee una estabilidad única, es resistente al calor y posee un peso molecular entre 30.000 - 400.000 p.m.
Los receptores mu representan el 22% de los R. opioides
Los receptores delta el 35%
Los receptores kappa el 42%
entre los tres reúnen el 99% de los receptores
Hoy se sabe que existen receptores diferentes, presentes en una misma célula, lo que se llama CONEUROTRANSMISIÓN.
Fuente:
RECEPTORES OPIOIDES MULTIPLES
Ya conocemos los 3 básicos, pero existe uno experimental pero tiene indicios de ser un receptor importante, dentro del receptor Mú hay subtipos (Mú1, Mú2), Kappa (k1, k2, k3), y Delta (Delta1y Delta2), el receptor en fase experimental se denomina receptor epsilon (E).
La analgesia está relacionada con receptores Mú básicamente de localización supraespinal, y Kappa en médula espinal, en otras palabrasa nivel supratentorio tenemos Receptores Mú y en espinal tenemos Kappa ambos modulan dolor, pero existen los haces espinotalámicos laterales por donde corre la vía del dolor y estos pasan obligatoriamente por médula para integrarse en los núcleos centrales tálamo y corteza por que si no el dolor se está presentando pero no integrándose por lo tanto no lo sentimos como tal, por que es una percepción subjetiva, no lo pueden medir para ser subjetivo tiene que integrarse en el cerebro.
Un compuesto opioide puede actuar en los receptores como agonista o antagonista dependiendo de donde esté y dependiendo del receptor que sea, ese opioide puede ser agonista aliviando el dolor, o como antagonista potenciándolo, los agonistas opioides tienen predilección por receptores Mú, los antogonistas como naloxona, otros que actúan como agonistas y antagonistas (nalofina, pentasufina), y aquellos que tienen efecto eufarizante o disforía (alteración del estado de ánimo), son poco antagonizado por naloxona, los fármacos de acción mixta (agonista y antagonista) pueden potenciar la acción de la naloxona y no eliminarla, los agonistas parciales como la Butrenolfina que tiene un efecto total sobre los receptores. Recordando lo que es la vía del dolor actúa sobre receptores que están a nivel de piel, estos a su vez transmiten por vías aferente por las fibras mielinizadas “A” o fibras desmielinizadas “C” entran por las raices dorsales y llegan al haz espinotalámicos laterales (dolor y temperatura), y también entramos al espinoreticular (dolor y tacto), que llega a formación reticular en médula y en meséncéfalo, tálamo y corteza sensorial, luego nos devolvemos por mesencéfalo, astas dorsales y llegamos a la sustancia gelatinosa de Rolando que es quién emite la respuesta motora, los neurotransmisores involucrados Glutamato y Aspartato (Aminoácidos excitatorios), Neuropeptidos (Sustancia P, Somatostatina, VIP, Colecistoquinina) y los Neuromoduladores (GABA, Glicina, 5-HT).
Fuente:
Conocidos como "Narcóticos" (que significa adormecimiento), se utilizan principalmente para combatir el dolor. Son legales para uso médico, por sus propiedades analgésicas.
Son derivados preparados a partir de la Goma de Opio (Papaver Somniferum), misma que se obtiene a partir del jugo extraído de los bulbos de la amapola.
En 1803 se aísla un alcaloide del opio al que se le llamó Morfina por el Dios griego del sueño "Morfeo", que es diez veces más potente que el opio y posteriormente se desarrollaron otros derivados como la Codeína que deprime la tos y la Heroína (1874), que es diez veces más potente que la morfina. Su nombre viene por ser la droga "heroica" en las guerras.
Los opiáceos son las drogas con mayor poder adictivo, debido a entran en el cerebro rápidamente. Entre los efectos que producen estas drogas están el de analgesia, somnolencia, cambios del estado de ánimo, depresión respiratoria, nausea, vómito, "miosis" (constricción pupilar) y disminución de la motilidad del tubo digestivo.
El efecto analgésico que produce la morfina tiene la particularidad de que ocurre sin pérdida de la conciencia y no afecta otras modalidades sensoriales, sin embargo es muy importante en caso de intoxicación, no permitir que el sujeto duerma ya que el efecto como depresor en la respiración, se suma al mismo efecto causado por el sueño y esto aumenta el riesgo de muerte por asfixia.
Los cambios conductuales se identifican con la euforia inicial, la apatía, lentitud psicomotora, el deterioro en el proceso del pensamiento y en la capacidad de atención y memoria, así como con cambios drásticos en actitudes escolares y sociales.
Fuente:
Opioides endógenos
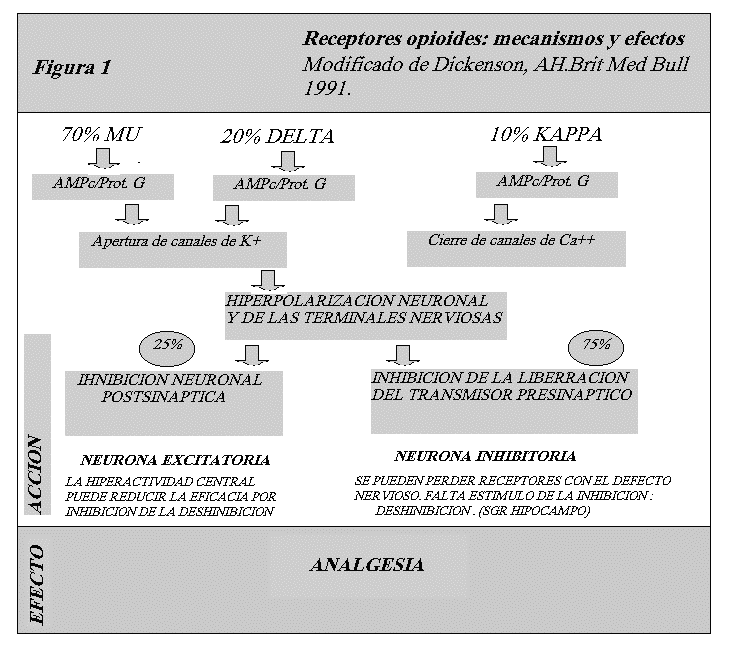
Existen tres grupos diferentes de opioides endógenos: encefalinas, endorfinas y dinorfinas. Los tres derivan de moléculas polipeptídeas precursoras: proencefalina, pro-opiomelanocortina y prodinorfina. En el SNC existen tres receptores opioides: mu, kapa y delta. La mayor concentración de receptores opioides se encuentra en las regiones sensoriales, hipotálamo, y sistema límbico del cerebro, en especial en la substancia gris periacueductal, y la amígdala. Los receptores opioides facilitan la neurotransmisión inhibitoria en el cerebro y en el sistema gastrointestinal. Inhiben la liberación de otros neurotransmisores y neuropéptidos como son dopamina, acetilcolina, 5HT y CRH. Estos receptores actúan todos uniéndose a la proteína G, y además el mu y delta actúan a través de los canales de potasio y la adenilciclasa, mientras que los kapa lo hacen inhibiendo los canales de calcio.
Fuentes:
ANTIVICIO:
ANTIALCOHÓLICOS:
Incluye tanto antiopiáceos como aversivos (bloqueo de la enzima acetaldehído deshidrogenasa)
ANTITABÁQUICOS:
Grupo que incluye:
Antagonistas opiáceos que actúan por competición específica con los receptores localizados principalmente en el sistema nervioso central, periférico , como la Naltrexona (antimu).
Sistema Opioide Endógeno y otras Drogas
En relación con este último punto se han obtenido en los últimos años evidencias claras de que el funcionalismo del sistema opioide endógeno no solamente está implicado en el abuso de sustancias como la heroína, sino que también juega un papel importante en las propiedades reforzadoras de otras drogas. De forma resumida podríamos decir que se ha propuesto la existencia de un sistema opioide endógeno de recompensa que sería activado por las drogas de abuso, de forma que al menos parte del potencial adictivo de las mismas podría modularse farmacológicamente mediante la utilización de fármacos activos sobre receptores opioides. Existen evidencias preclínicas de que ésto podría ser así en el caso de los psicoestimulantes, cannabinoides, nicotina y alcohol; con esta última droga de abuso se ha podido demostrar la utilidad clínica de esta estrategia terapéutica, ya que es bien conocida la disminución del consumo de alcohol que puede obtenerse en pacientes tratados con el antagonista opioide naltrexona. No sería extraño por tanto que en un futuro nos encontremos con una utilización más extendida de ligandos opioides (posiblemente agonistas parciales o antagonistas) en tratamientos de deshabituación a diversas drogas de abuso, o bien con sustancias activas sobre el sistema opioide endógeno en virtud de otros mecanismos de actuación. La vulnerabilidad individual al consumo de drogas es otro de los fenómenos en los que el sistema opioide endógeno parece estar significativamente implicado. Trabajos realizados con cepas de ratas especialmente susceptibles al efecto reforzador de distintas drogas muestran que estos animales exhiben diferencias en cuanto a su tono opioide basal o a la respuesta del sistema opioide tras la exposición a una droga. También en el caso del alcohol nos encontramos con datos clínicos concordantes, ya que se ha demostrado que la predisposición genética al consumo de dicha sustancia se correlaciona positivamente con el incremento que produce sobre la liberación de b-endorfina y encefalinas. Conclusión Se puede concluir por tanto que el estado actual de la investigaciones sobre el papel del sistema opioide endógeno en las drogodependencias abre nuevas expectativas farmacológicas para ayudar a limitar el consumo de distintas drogas de abuso, de forma semejante a lo ya descrito en el caso de la naltrexona y el alcohol.
Fuente:
En el caso de los antitabáquicos ver antidepresivos inhibidores de la recaptación neuronal de noradrenalina y dopamina como el Bupropion en los antitabáquicos.
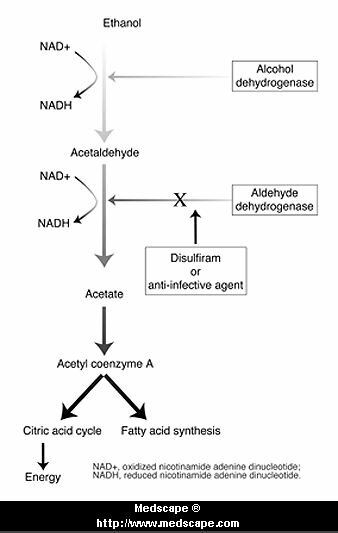
En el caso del Disulfiram:
Inhibe la oxidación del acetaldehído (producto del metabolismo del alcohol) y es por ello que la ingestión de alcohol durante el tratamiento con disulfiram provoca una molesta y desagradable respuesta (vómitos, cefaleas, disnea, sudoración, precordialgias).
Su mecanismo de acción se debe a la inhibición de la aldehído deshidrogenasa hepática. El acetaldehído es responsable de los efectos desagradables que persisten hasta tanto se metabolice el alcohol ingerido sin interferir su eliminación. El disulfiram se absorbe y elimina lentamente; los efectos se conservan hasta una o dos semanas después de ingerida la última dosis; no produce tolerancia.
COMENTARIOS DE: Nalbufina
POR FAVOR NECESITO TODA ESTA INFORMACION, PARA TRABAJO DE LA UNIVERSIDAD Y EN ESTA PAGINA ESTA TODO PERO NO PUEDO INPRIMIRLO. NOMBRE GENERICO /DOSIS DEL MEDICAMENTO/ VIA ORAL ,INYECTABLE. PIEL/MEDICAMENTO COMPATIBLES Y NO COMPATIBLES/ EN QUE SE PUEDEN DILUIR Y EN QUE NO SE PUEDEN DILUIR/ INDICACIONES Y CONTRA INDICACIONES. GRACIAS
Comentarios 1 a 1 de 1
6 meses para responder posts antiguos
¡Queremos tu opinión!
Si pregunta sobre este fármaco revise antes estos dos enlaces en particular
Ahora ya puede entender el prospecto y la ficha técnica